Command Palette
Search for a command to run...
Scientists Have Independently Generated Novel Materials by reverse-engineering gallium-containing Materials Using a Bayesian Optimization framework. The Optimization Results Exhibit Uniqueness and novelty.

In the modern semiconductor industry, the boundaries of material performance are constantly being pushed to higher dimensions. From high-efficiency photovoltaic devices to high-brightness light-emitting diodes (LEDs), and then to high-frequency communication and quantum information systems, almost all key technologies rely on a core capability at their core—Precise control over the electronic structure of materials, especially the precise design of the band gap.However, this goal has long been difficult to achieve in the traditional materials science system.
The reason is that the electronic properties of a material are not simply determined by a single element, but are influenced by complex chemical bonding, crystal structure, electron orbital hybridization, and the synergistic effects of multiple elements. Among many material systems, gallium-based semiconductors occupy a unique position.Gallium's excellent chemical diversity and multivalence state characteristics enable it to exhibit a range of tunable electronic properties, from wide bandgap to narrow bandgap.
Gallium-containing compounds have become a crucial foundation for key optoelectronic and energy conversion technologies such as high-efficiency solar cells, high-brightness LEDs, and high-frequency communication devices. They are also emerging as potential candidate materials for flexible, biocompatible, and implantable electronic systems. However, despite decades of research, the discovery of novel gallium-containing materials targeting specific electronic properties still largely relies on empirical exploration.This is mainly limited by the vast design space of components and the high computational cost of calculations based on first principles.
Against this backdrop, a research team led by Flinders University in collaboration with Khalifa University in the UAE has proposed a machine learning-guided Bayesian optimization (BO) framework that enables the reverse design of gallium-based components with predefined electronic properties while maintaining chemical rationality.
With the help of this unified framework,The system can autonomously generate novel, chemically effective gallium-containing materials and achieve a tunable bandgap of 0.5–3.5 eV.This energy range is of great significance for applications in solar energy, photonics, and power electronics. The Bayesian optimization process can adaptively guide the search to the region with the highest "desired improvement". The optimized analysis results show that the generated material has 100% uniqueness and novelty relative to the training data, and the SMACT effectiveness is significantly improved in the 1.5–2.5 eV bandgap range.
The related research findings, titled "Bayesian Optimization-Guided Discovery of Gallium-Containing Semiconductors with Targeted Band Gaps," have been published in ACS Publications.
Research highlights:
The new framework can accelerate inverse material design under realistic chemical constraints, providing an alternative to traditional screening methods based on DFT (density functional theory).
* The new framework not only efficiently covers chemically plausible regions, but also maintains a high degree of novelty and component diversity compared to existing databases.
* This research breaks through the limitations of traditional static property prediction, propelling semiconductor discovery towards a data-driven generative research paradigm.

Paper address:
https://pubs.acs.org/doi/10.1021/acsmaterialslett.5c01482
Datasets: Constructing a chemical learning space from real-world materials databases
This study used the NOMAD and Materials Project databases to train the model.The data includes the chemical composition of the material and its corresponding experimental band gap value.For example, Ga₄P₄, GaAs, GaN, Ga₂O₃, etc. The initial dataset contains 2,530 material compositions and their band gap records.
To ensure data quality, samples with missing values in the "composition" or "band_gap" columns were removed. Non-physical or negative bandgap data were also eliminated, and duplicate records were removed, ultimately retaining 1,578 valid components for modeling. Furthermore, the chemical formula strings were standardized using the pymatgen package to merge chemically equivalent terms. The bandgap unit was uniformly converted from Joules to electron volts (eV). In the preprocessed dataset, the bandgap ranged from 0.0 to 5.92 eV, with a mean of approximately 1.8 eV and a standard deviation of 1.6 eV.
The study further screened the material composition, retaining only compounds containing elements from a predefined set of atomic numbers to ensure the research focused on gallium-based material systems. Several additional features were also constructed, including:
* Number of elements in each chemical formula
* Length of chemical formula string
* A binary indicator of the presence or absence of gallium
The dataset was then randomly divided into training and test sets in an 8:2 ratio, with the split performed at the "component level" to avoid chemically similar compounds appearing in different datasets simultaneously. Five-fold cross-validation was also employed to evaluate the model's robustness under different data partitioning conditions.
Framework: Co-design of Machine Learning and Bayesian Optimization
This study proposes a chemically constrained Bayesian optimization (BO) framework.As shown in the figure below, it first uses a gradient boosting regression model trained on a gallium-based composite material dataset to predict the band gap of the material; then, Bayesian optimization is used to iteratively explore in a constrained composition space; finally, the generated candidate materials are screened for chemical validity, novelty and uniqueness using SMACT and pymatgen tools, thereby identifying gallium-based composite materials with the best performance that have not been explored before.
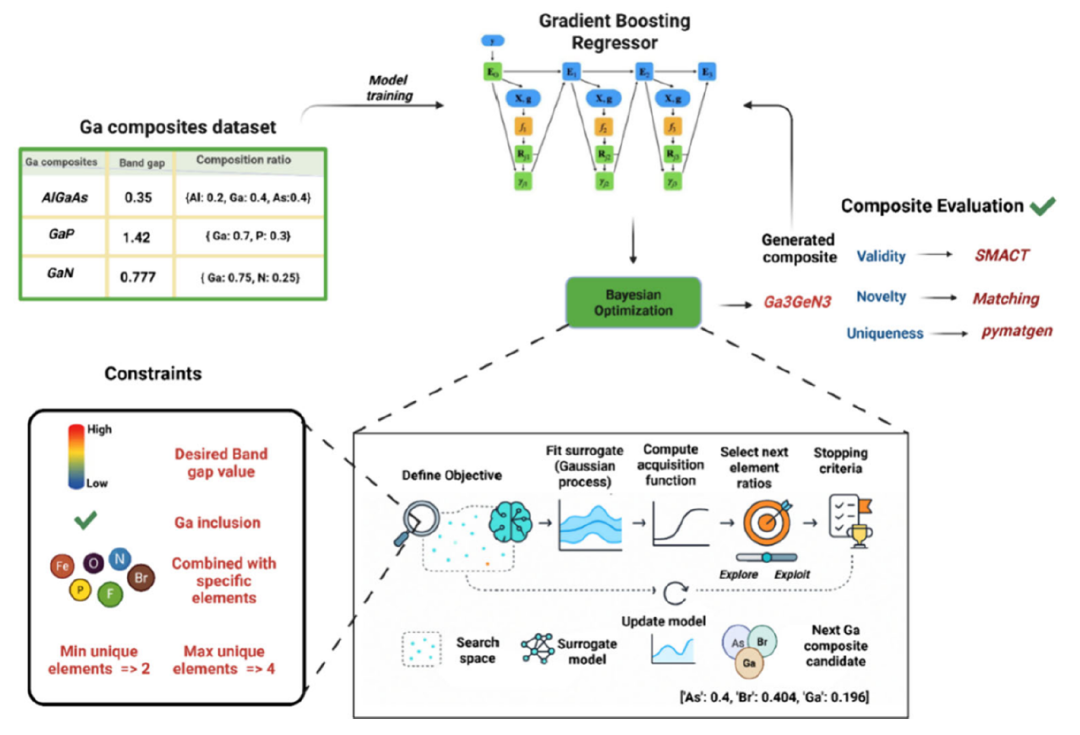
Prediction Model Layer
This study systematically evaluated eight machine learning regression algorithms, including linear models, support vector regression, random forests, gradient boosting, and K-nearest neighbors (KNN). The results show that the nonlinear model significantly outperforms the linear model overall, indicating a strong nonlinear relationship between material composition and band gap.The KNN model performed best, achieving an R² of 0.812, and also outperformed other models in terms of error metrics.
Of all the candidate models, KNN was ultimately selected as the surrogate model in Bayesian optimization.The reason is that it has excellent local interpolation capabilities and maintains stable performance under different random partitioning conditions.Unlike tree-based ensemble models, KNN can preserve neighborhood relationships in the component feature space, which is crucial for identifying similarities between materials with similar proportions of elements.
In Bayesian optimization scenarios, this "local preservation capability" is particularly important because optimization searches often focus on potential regions near known high-quality candidates. Therefore, the non-parametric and locally adaptive characteristics of KNN can provide the optimizer with smoother and more reliable search guidance, while maintaining high computational efficiency in sparsely sampled material spaces.
Bayesian Optimization module
This BO workflow utilizes the KNN surrogate model to guide the search for gallium-containing components in the target bandgap.By employing the "Expected Improvement" acquisition function, a balance is struck between "exploration" and "utilization," thereby generating candidate stoichiometry in a gallium-centric composition space.
The system sets several constraints, including: each component must contain a maximum of 4 elements and must meet a minimum gallium content requirement to ensure that candidate materials remain relevant to gallium-based research topics.
Chemically constrained filter layer
All generated candidate materials must be verified using the SMACT tool, including constraints such as charge balance, reasonable oxidation state, and electronegativity consistency, to ensure that the generated materials are not only valid in mathematical space but also chemically realizable.
Furthermore, the framework incorporates Explainable Artificial Intelligence (XAI) methods, utilizing SHAP to analyze model decision logic, thus transforming material prediction from a "black box" to an "explainable system".
Accelerating reverse materials design under realistic chemical constraints
Researchers designed a series of experiments to evaluate and analyze the model's performance, structural features, interpretability, and chemical validity:
Model performance evaluation
In terms of model performance evaluation, the KNN model showed stability in cross-validation, with an R² of approximately 0.60 ± 0.07 and an RMSE of approximately 1.02 eV, indicating that the model has good generalization ability in sparse chemical spaces.
As shown in the feature importance analysis below, melting point, electronegativity range, and electronegativity deviation are key factors affecting band gap prediction, which are closely related to bond strength and charge transfer behavior in materials. As electronegativity differences increase, the band gap tends to decrease, while increases in melting point and cohesive energy correspond to larger band gaps—a pattern highly consistent with traditional semiconductor physics.
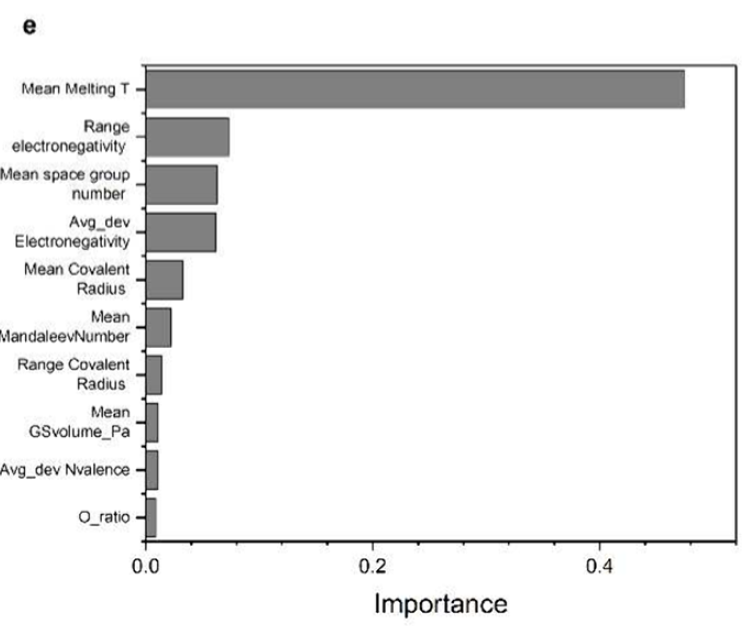
The ability to learn real chemical rules from data
During the generation phase, Bayesian optimization proposed 1,025 candidate gallium-containing components, of which only 38 passed the SMACT screening, indicating that the chemical feasibility constraints were extremely strict.These effective materials are mainly concentrated in the 2.0–2.5 eV range, which means that it is easier to form medium-bandgap semiconductors with both ionic and covalent bond characteristics in this region. These results are highly consistent with known systems, such as Ga₂O₃ (≈4.8 eV) and Ga₂S₃ (≈2.5 eV).
The BO search process also shows a tendency to cluster towards known gallium-containing chemical families (such as Ga–O, Ga–N, Ga–As/Sb), and proposes new intermediate stoichiometry in these regions, such as: Ga₀.₅₁As₀.₁₆N₀.₂₄Sb₀.₁₀, Ga₀.₁₇₁Sb₀.₁₇₅O₀.₃₆₇F₀.₂₈₆.
For wide bandgap materials (>3.0 eV), the algorithm favors oxygen-rich compounds because strong Ga–O bonds help widen the bandgap; while lower bandgap materials (around 1.5–2.0 eV) are typically achieved by replacing oxygen with sulfur, selenium, or phosphorus, introducing stronger p–p interactions. These patterns are highly consistent with existing experimental observations, indicating that the model has been able to "implicitly learn" real chemical rules from the data.
The ability to capture real-world "structure-property relationships"
To confirm that the generated gallium-containing composition corresponds to a "physically realizable" material, the research team used the Chemelon-dng model developed by Park et al. to predict its crystal prototype, as shown in the figure below:
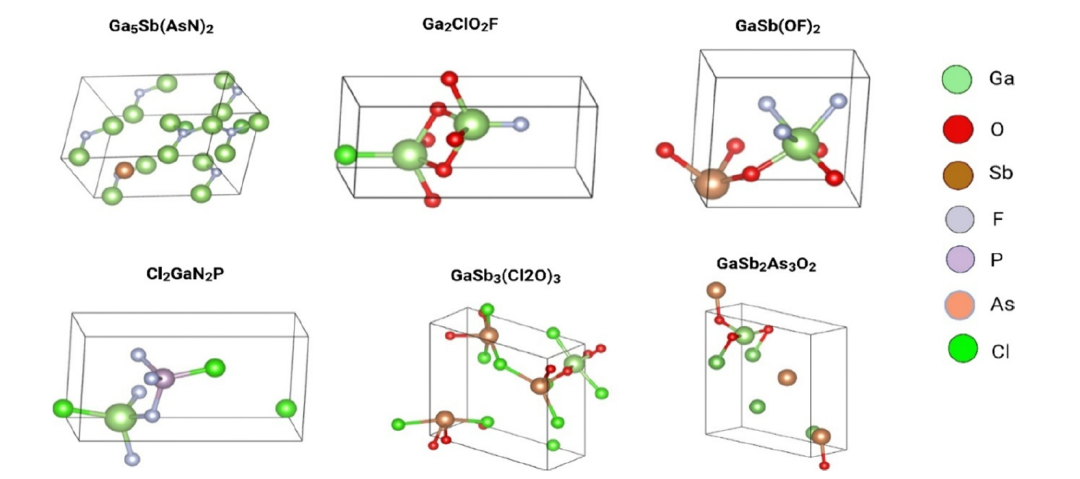
The candidate components validated by SMACT exhibited chemically reasonable coordination environments, dominated by tetrahedral and octahedral gallium centers, which is highly consistent with known crystal prototypes such as Ga₂O₃, GaN, and GaSe. The surrogate model successfully reproduced the empirically consistent electronic structure hierarchy relationships: oxides: 3.5–4.8 eV, chalcogenides: 1.8–2.6 eV, and group A nitrogen compounds: approximately 1.2–2.0 eV, i.e., oxide band gap > chalcogenide band gap > group A nitrogen band gap.
This result indicates that,This Bayesian optimization workflow is now able to effectively capture real-world "structure-property relationships".
It is worth noting that none of the 38 valid components that were verified were duplicates of existing known materials, which further proves that the generated results have both "novelty" and "chemical consistency".
DFT verification
The researchers further conducted DFT validation. The table below summarizes the comparison results of "model-predicted bandgap" and "DFT-calculated bandgap" among the 10 components that passed SMACT validation, as well as the corresponding bandgap types.
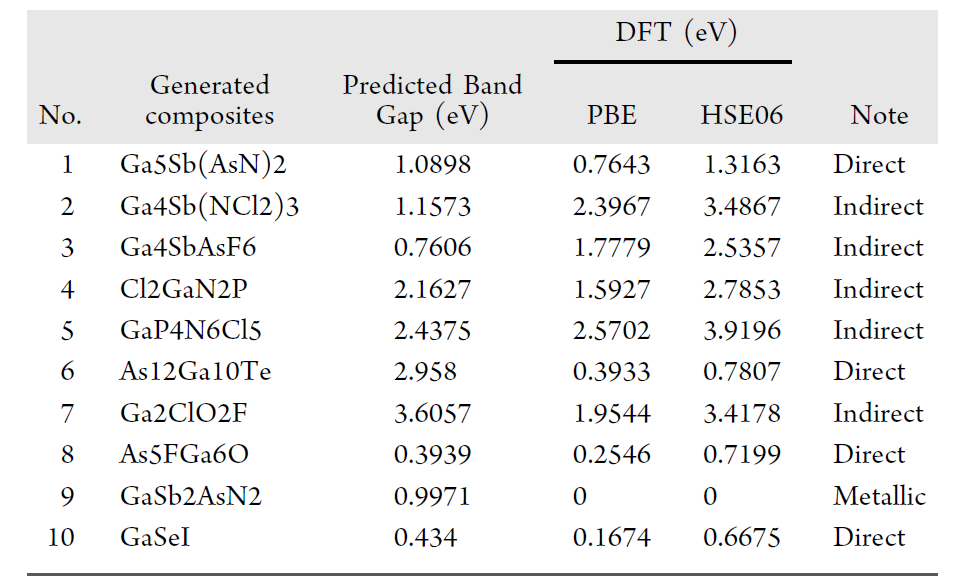
Overall, the mean absolute error (MAE) was 0.890 eV, the root mean square error (RMSE) was 1.158 eV, and the median absolute error was 0.784 eV. Although there are some biases, it has high practical value in the early screening stage of material discovery. More importantly, all the validated materials were not found in known databases, demonstrating high novelty.
Conclusion
Overall, this study demonstrates a novel material design paradigm for gallium-containing semiconductors: an automated generation path from "data" to "new materials" through the synergistic effect of machine learning modeling, Bayesian optimization search, and chemical constraint screening.
From an industry perspective, this approach has potential value for photovoltaic material design, light-emitting device development, and wide-bandgap semiconductor research. Especially against the backdrop of the rapid development of next-generation power electronics and optoelectronic devices, the demand for bandgap-controllable materials is growing rapidly, and AI-driven material design methods are expected to become a key tool to accelerate this process.
Furthermore, the significance of this framework is not limited to the gallium system; its methodology can also be extended to indium, tin, and even lead-free semiconductor systems, providing a general path for the rational design of complex multi-component compounds. This marks a new stage in materials science, moving from "experience-based trial and error" to "algorithm-based generation," with artificial intelligence becoming a core bridge connecting chemical rules and materials discovery.
References:
https://techxplore.com/news/2026-05-ai-discovery-gen-chips-electronic.html
https://pubs.acs.org/doi/10.1021/acsmaterialslett.5c01482








