Command Palette
Search for a command to run...
Reshaping the Predictive Power of Disordered Protein Assemblies, NVIDIA, MIT, Oxford University, University of Copenhagen, Peptone, and Others Release Generative Models and New benchmarks.

In the history of structural biology, the principle that "structure determines function" was once considered an almost unshakeable fundamental law. Both the classic helical conformation of insulin and the tetrameric structure of hemoglobin reinforced the consensus that proteins must possess a stable three-dimensional structure to perform their biological functions.
However,The discovery of intrinsically disordered proteins (IDPs) and their intrinsically disordered regions (IDRs)This traditional understanding is being constantly reshaped. They do not form fixed structures under physiological conditions, but they are deeply involved in core processes such as signal transduction and gene transcription regulation, and are closely related to major human diseases such as cancer and neurodegenerative diseases.
Computational biology research has further revealed that approximately 30% amino acid residues in the eukaryotic proteome are in a disordered state. This means that disorder is not "abnormal," but rather a normal component of living systems. However,The highly dynamic nature of disordered proteins makes them difficult to capture stably using traditional experimental techniques, and also difficult to accurately simulate their conformational distribution using conventional computational methods.This has become a long-standing technological bottleneck in the field.
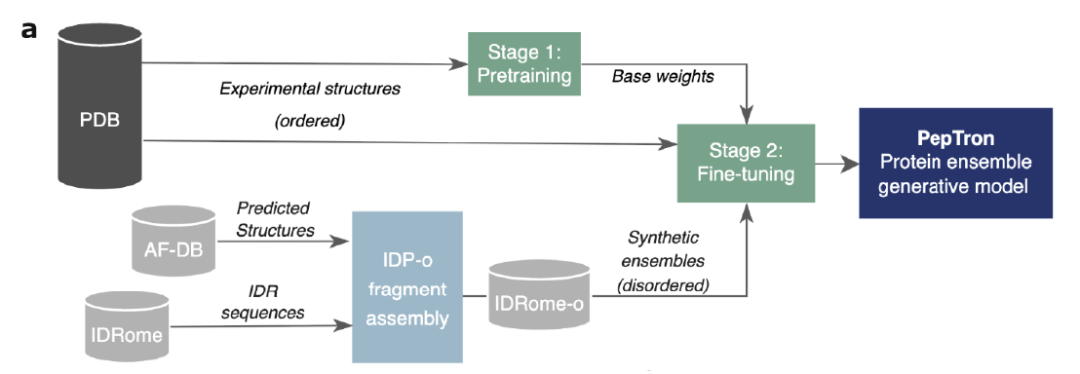
To address this challenge, a joint team comprised of Peptone, a UK-based protein analysis technology developer, the University of Copenhagen, NVIDIA, the University of Oxford, MIT, Duke University, and others has proposed two key breakthroughs.One is the PeptoneBench system evaluation framework.This framework integrates multi-source experimental data such as SAXS, NMR, RDC, and PRE, and combines statistical methods such as maximum entropy reweighting to achieve a rigorous quantitative comparison between experimental observations and theoretical predictions.The second is the generative model PepTron.Training on an expanded synthetic IDR dataset specifically enhances the ability to model disordered regions, enabling it to better capture the conformational diversity of disordered proteins.
The research team used PeptoneBench to systematically compare PepTron with mainstream prediction tools such as AlphaFold2, Boltz2, and BioEmu. The results showed that PepTron demonstrated a high degree of consistency with experimental results in predicting both ordered and disordered regions, reaching state-of-the-art (SOTA) performance. Based on these advances, a more accurate and biologically realistic framework for predicting protein structures using a "conformation set" is emerging, significantly improving our overall understanding of proteins across their entire ordered-disorder spectrum.
The related research findings, titled "Advancing Protein Ensemble Predictions Across the Order–Disorder Continuum," have been published as a preprint on bioRxiv.

Paper address:
https://www.biorxiv.org/content/10.1101/2025.10.18.680935v1
Follow our official WeChat account and reply "PepTron" in the background to get the full PDF.
More AI frontier papers:
https://hyper.ai/papers
Systematic Construction of PeptoneBench and Multi-Source Experimental Datasets
Protein databases (PDBs) are the most fundamental and important public resources in structural biology, but there are significant structural gaps in their coverage of intrinsically disordered proteins (IDPs) and their disordered regions (IDRs).Only about 3% entries were marked as unordered.In the human proteome, however, the proportion of such disordered regions is as high as 20–30%.
As shown in the figure below, this systematic bias causes most structural prediction models to naturally "prefer" stable conformations, limiting their ability to learn from dynamic, disordered states in the long run. To compensate for this deficiency,Researchers introduced supplementary databases such as IDRome, which have an unordered proportion of approximately 771 TP3T.It can complement the PDB in terms of statistical distribution. However, this database lacks structural data analyzed in real experiments, making it difficult to use as a direct benchmark for modeling and evaluation, and its application value is still significantly limited.
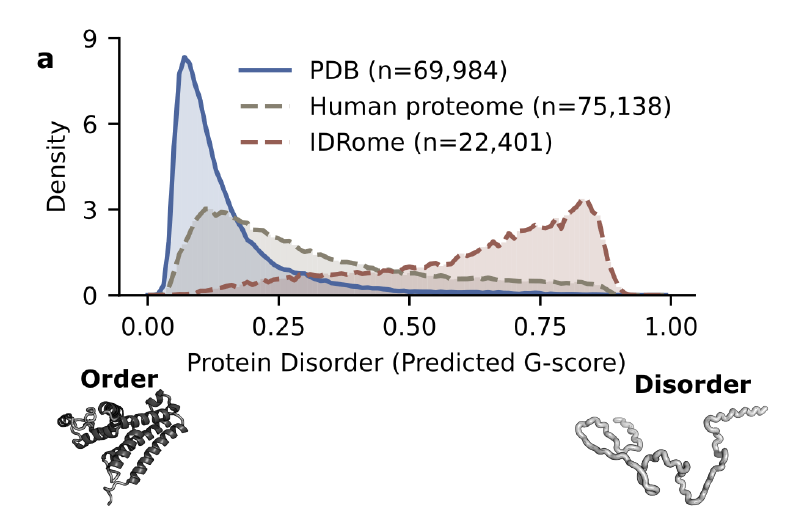
To overcome the aforementioned data bottlenecksThe first step is to establish quantifiable and comparable indicators of disorder.This study uses the average G score of proteins as the core metric, with values ranging from 0 (completely ordered) to 1 (completely disordered). Calculated based on NMR secondary chemical shift (CS) data, it accurately reflects the tendency for local secondary structure formation. For proteins lacking experimental CS data, the research team used the ADOPT2 machine learning model trained on TriZOD to predict the G score, thus achieving a unified quantification of the entire order-disorder spectrum.
Building on this, the team further pointed out that relying solely on structural data from PDBs cannot objectively assess the quality of conformation sets.Therefore, it is necessary to construct an experimental dataset that covers the complete ordered-unordered range.
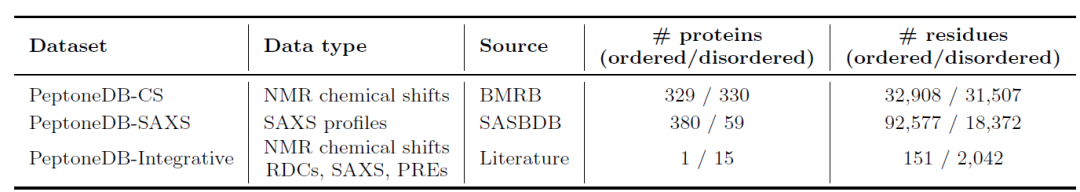
To this end, as shown in the table below, researchers established three complementary data resources: PeptoneDB-CS (NMR chemical shifts derived from BMRB), PeptoneDB-SAXS (SAXS spectra from SASBDB), and PeptoneDB-Integrative (a dedicated IDP set integrating multiple orthogonal experimental data). These three types of data have different structures and complementary information: CS reveals local structures, SAXS reflects the overall conformation, and Integrative supports cross-validation.
Based on this data, as shown in the figure below.Researchers developed the PeptoneBench evaluation framework to quantify the consistency between the predicted conformation set and experimental data.The entire process includes: conformation set standardization and preprocessing; mapping the predicted structure to observations comparable to experiments using a forward model; consistency scoring based on normalized RMSE, incorporating uncertainties from both the model and experiments throughout the process. The final results are presented as an RMSE–G score graph, and errors are estimated using Lowes smoothing and bootstrapping, further synthesized into a PeptoneBench aggregate score, forming a quantitative standard for directly comparing the performance of different tools.
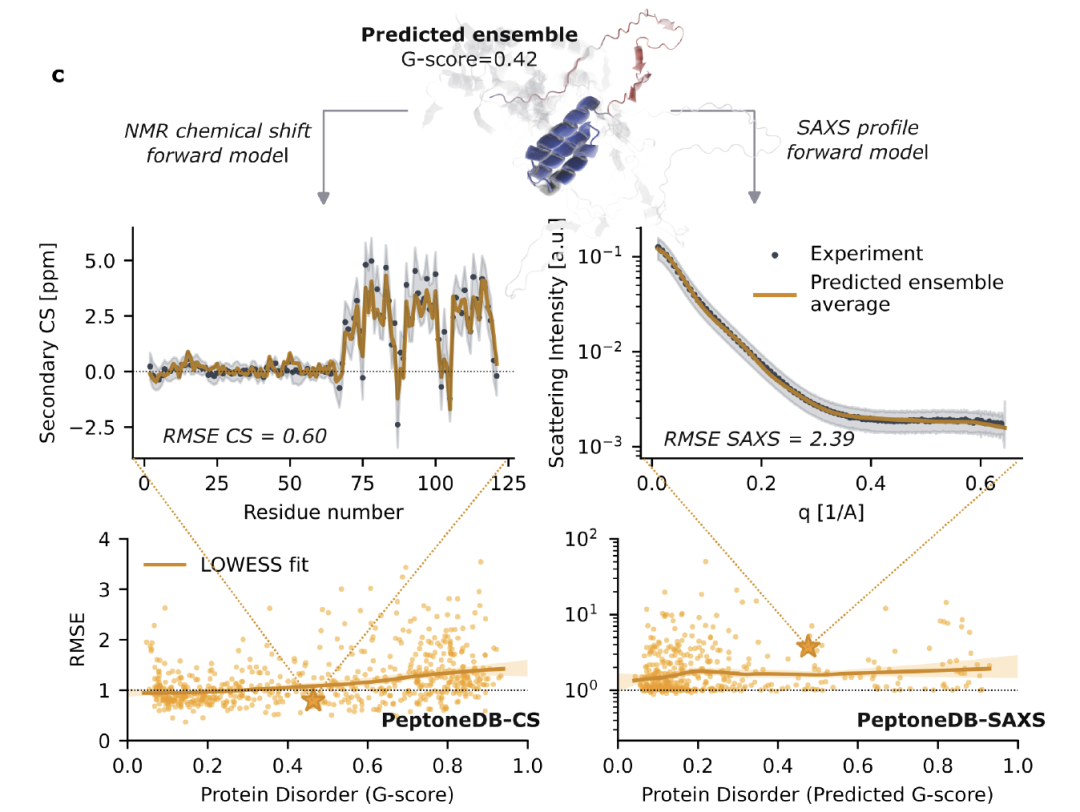
It is worth emphasizing that some initial sets of conformations with high RMSE may actually be closer to the experimental distribution after being reweighted using maximum entropy. To avoid misjudging "incorrect weights" as "missing conformations,"PeptoneBench also reports the RMSE before and after reweighting to distinguish between correctable sampling bias and irrecoverable conformational loss.This strategy is particularly crucial for IDPs, which are highly dynamic and extremely sensitive to experimental conditions: as long as the generative model can cover a sufficiently rich conformational space, it can quickly adapt through the reweighting process even if the experimental environment is different, thereby significantly improving the practicality and reliability of the prediction results.
PepTron: A conformational model that balances ordered and disordered proteins
The proposed PepTron model is a protein conformation generator built on the ESMFlow flow matching architecture. Its goal is to cover the complete conformation spectrum from completely ordered to highly disordered, generating a set of conformations that are both physically plausible and structurally diverse.
In terms of model architecture,PepTron is based on ESMFlow and implemented in the NVIDIA BioNeMo framework to improve training and inference efficiency.The model integrates the cuEquivariance triangular attention mechanism and supports flow matching functionality through BioNeMo's Modular Co-Design subpackage. The training process follows BioNeMo's distributed best practices, combining multiple parallel strategies and mixed-precision computation, thus enabling stable and efficient scaling in multi-GPU environments.
It is worth emphasizing that PepTron does not rely on multiple sequence alignment (MSA) or external ESM weights during the inference phase. It can generate a complete set of conformations with only a single checkpoint, which greatly simplifies the threshold for use.
To address the challenge of scarce experimental structure data in disordered regions, the research team constructed a synthetic structure dataset, IDRome-o, based on IDRome. Therefore,They developed IDP-o, a fragment assembly-based protein structure generation tool that can generate physically plausible sets of IDP conformations on a large scale at extremely low cost. IDP-o combines fragment assembly and hierarchical chain growth strategies to extract six-residue fragments from the AlphaFold database, which contains 214 million structures, thereby more accurately capturing transient helical structures in disordered proteins.
It should be noted that the goal of IDR-o is not to simulate a certain equilibrium distribution, but to cover all reasonable conformations that the sequence may sample. Therefore, its output is particularly suitable for subsequent maximum entropy reweighting and can also serve as a high-quality initial conformation library for molecular dynamics simulations.
To overcome the bias of traditional models that tend to predict stable structures, as shown in the figure below,PepTron employs a hybrid training strategy combining experimental data and synthetic data.First, the model is pre-trained using the experimentally resolved structures from the PDB database. Then, a synthetically generated set of disordered proteins is introduced for fine-tuning, allowing the model to fully learn the continuous distribution of ordered and disordered conformations. Even under computationally limited conditions, this strategy significantly improves the model's predictive performance on various proteins.
In terms of specific training procedures,The research was divided into two phases:In the initial stage, starting with ESMFold weights, the flow matching module is retrained using PDB data, and the sequence length trimming range is expanded to 512 residues. In the hybrid fine-tuning stage, a hybrid dataset consisting of PDB experimental structures and IDRome-o synthetic data is used as training data to perform final optimization of the model. This design enables PepTron to access the entire ordered-disorder spectrum, achieving a more comprehensive and realistic modeling of the dynamic conformational space of proteins.
Model Validation for Full-Spectrum Conformations: A Systematic Comparison of PepTron and Mainstream Methods
The research team then used the PeptoneBench framework to systematically evaluate PepTron's performance on experimental data completely independent of the training set, and compared it with mainstream models such as ESMFold, ESMFlow, AlphaFold2, Boltz2, and BioEmu. Simultaneously, the team conducted specific tests on the PeptoneDB-Integrative dataset, which focuses on intrinsically disordered proteins (IDPs), to comprehensively examine the capabilities of each model in modeling disordered conformations. The results showed clear differentiation characteristics among the models.
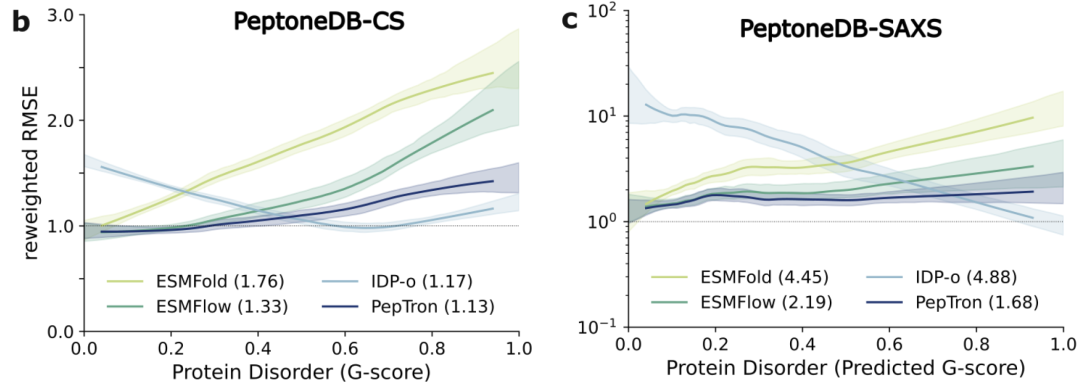
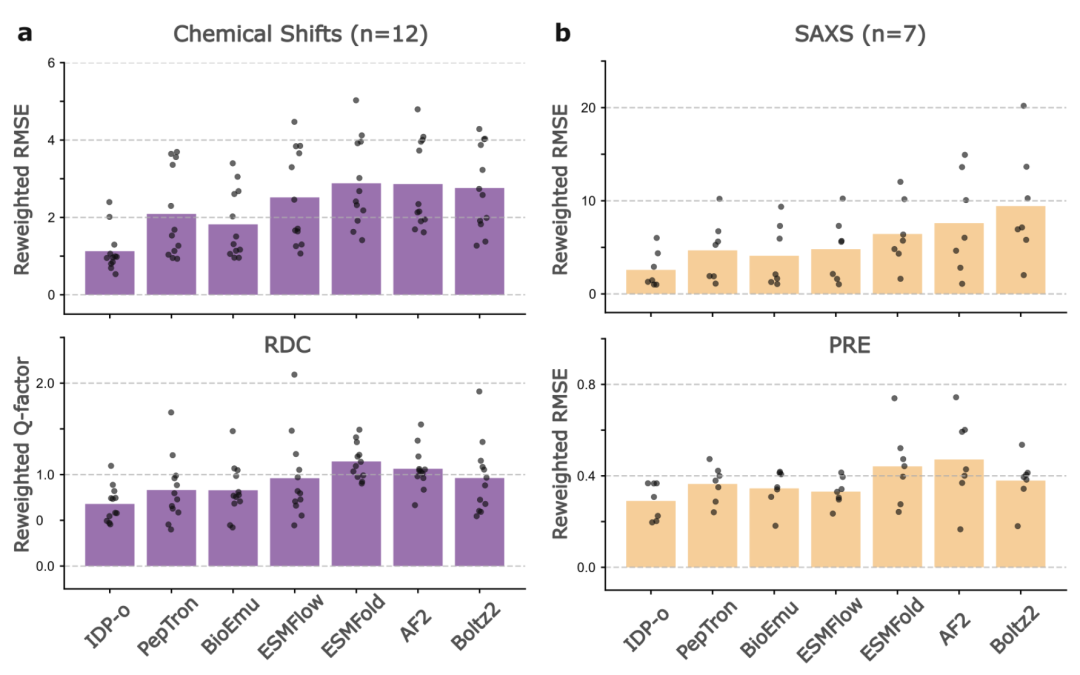
As shown in the figure below, on the PeptoneDB-CS dataset, the performance of each model varies significantly with the degree of protein disorder (G score): ESMFold and ESMFlow are accurate in predicting ordered regions, but their performance is significantly reduced in disordered regions; IDP-o shows a typical complementary pattern—the higher the degree of disorder, the better the performance.PepTron maintains a stable high consistency across the entire ordered-disorder conformation spectrum.This balancing ability was further validated in the PeptoneDB-SAXS dataset and subsequent reweighted analysis, demonstrating that PepTron can effectively capture the conformational diversity of disordered proteins without sacrificing the accuracy of ordered structures.
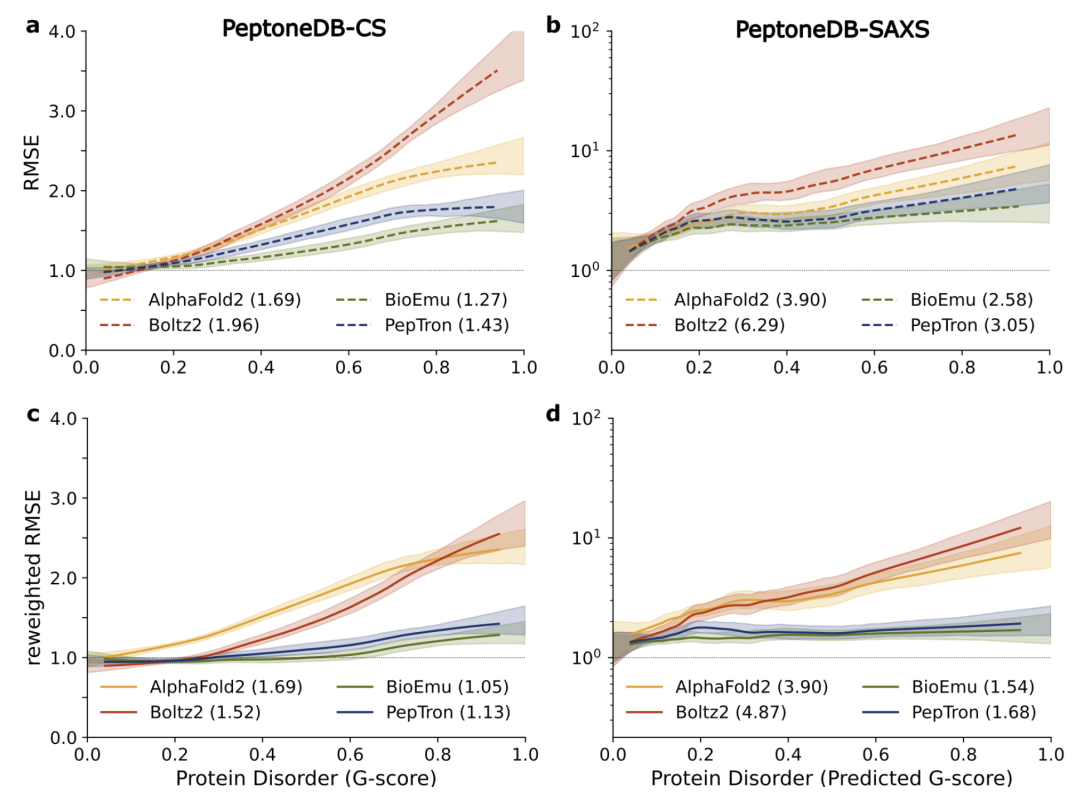
Further cross-model comparison results are shown in the figure below. Although AlphaFold2 and Boltz2 still dominate in predicting ordered proteins, their performance systematically declines as the degree of disorder increases; in contrast,PepTron and BioEmu exhibit stronger robustness across the entire conformational spectrum, making them more suitable for handling the highly heterogeneous structural features of IDPs.
To ensure that training on disordered regions did not impair its predictive ability for ordered proteins, the research team also conducted additional tests on ordered structure data from CAMEO22 and CASP14. The results showed that...PepTron performs in line with ESMFlow on key metrics such as RMSD, LDDT, and TM, demonstrating that it does not compromise the accuracy of ordered structures while extending IDR modeling capabilities.
In the PeptoneDB-Integrative dataset, which integrates multiple experimental metrics, as shown in the figure below, the model performance further reveals differences. IDP-o performs particularly well after maximum entropy reweighting, significantly outperforming other models in both RMSE and RDC Q factor; PepTron and BioEmu are similar in RDC metrics, but BioEmu is more advantageous in predicting local chemical shifts. It is worth noting that even under unweighted conditions,IDP-o continues to lead in most local and global metrics, demonstrating its natural advantage in covering disordered protein conformations.
The study also pointed out several common bottlenecks in the current model:Most models fail to capture long-range contact preferences and exhibit varying degrees of secondary structure bias. Furthermore, mainstream models generally struggle to accurately describe the unfolded state of "conditionally folded sequences," while IDP-o demonstrates unique superiority in this regard.
From Disorder to Order: Global Breakthroughs and New Chapters in IDP Research
Intrinsically disordered proteins (IDPs) are rapidly becoming a research frontier in the global life sciences and pharmaceutical industries due to their highly dynamic conformational characteristics and close association with many major diseases.
In academia, AI structure prediction technology is becoming a key force in cracking the "dynamic password" of IDPs.The AlphaFold-Metainference method proposed by Cambridge University,By combining AlphaFold alignment error maps with molecular dynamics simulations, this approach overcomes the limitation of traditional AlphaFold, which mainly predicts stable structures, and successfully constructs IDPs and sets of structures containing disordered regions, providing a new path for understanding their polymorphism.
Paper title:
AlphaFold prediction of structural ensembles of disordered proteins
Paper link:https://www.nature.com/articles/s41467-025-56572-9
The University of Copenhagen team further integrated AlphaFold with a protein language model.It has enabled large-scale prediction of the conformation of the human disordered proteome.This demonstrates the universality and scalability of AI technology in IDP research.
Paper title:
Conformational ensembles of the human intrinsically disordered proteome
Paper link:https://www.nature.com/articles/s41586-023-07004-5
Whether academic findings can truly change disease treatment depends on the industry's ability to translate technology into practical applications. The collaboration between British biotechnology company Peptone and German pharmaceutical company Evotec...This demonstrates a feasible path for extending IDP research into drug development.Leveraging Peptone's ultrafast hydrogen-deuterium exchange mass spectrometry (HDX-MS) platform, researchers can track the dynamic changes of disordered proteins in real time and capture binding sites that are difficult to identify using traditional structural determination methods. Combined with Evotec's advantages in target validation, drug screening, and clinical advancement, it is possible to transform the difficult-to-drug IDP target into a candidate molecule with drug potential.
This series of advancements not only echoes the trend of the PepTron model "covering the entire spectrum of ordered-disordered structures," but also signifies that disordered proteins, once considered elusive, are gradually becoming key targets in precision medicine and biopharmaceuticals. With continued technological breakthroughs and deepening industry collaboration, IDPs may provide a completely new framework for understanding and intervention pathways for future disease treatment.
Reference Links:
1.https://www.vbdata.cn/intelDetail/717834
2.https://c.m.163.com/news/a/JDIR2LQJ0552ZPM2.html
3.https://www.vbdata.cn/intelDetail/580634