Command Palette
Search for a command to run...
MIT Has Developed the Pichia-CLM Model to Learn the "language" of Yeast DNA, Potentially Increasing the Yield of Exogenous Proteins by up to Three times.

In the fields of biopharmaceuticals and industrial biotechnology, efficient expression of recombinant proteins remains a core factor determining production costs and process feasibility. From monoclonal antibodies and vaccine antigens to industrial enzyme preparations, even a slight increase in expression levels can bring significant economic value.
In many expression systems,Pichia pastoris (Komagataella phaffii) is valued for its high-density fermentation capacity, mature secretory expression system, and excellent protein processing capabilities.It has become one of the important hosts for industrial production. However, a long-standing problem that has plagued the industry is that even if the amino acid sequences are completely identical, simply changing the "synonymous codons" in the coding DNA can lead to orders of magnitude differences in expression levels.
This phenomenon stems from codon usage bias (CUB)—in many organisms, certain synonymous codons are preferentially used. The selection of synonymous codons affects protein yield by influencing transcription, mRNA stability, translation, protein folding, post-translational modifications (PTMs), and solubility.Therefore, "codon optimization" has become a key step in the expression of exogenous proteins.
Currently, various codon optimization tools and methods based on host CUBs have been developed in the industry, but these methods may still not consistently produce highly expressive constructs. In recent years, with the development of artificial intelligence, especially sequence modeling techniques,Researchers have begun to view gene sequences as a kind of "language" and are trying to learn the implicit rules within them using methods similar to natural language processing.
In this context,A research team from MIT has proposed a deep learning-based language model, Pichia-CLM, for codon optimization in the industrial host Pichia pastoris to improve the yield of recombinant proteins.Unlike traditional methods that rely on CUB metrics (which typically only provide a global score and ignore sequence context), Pichia-CLM utilizes host genome data to learn the amino acid-to-codon mapping unbiasedly. Researchers experimentally validated Pichia-CLM on six protein classes of varying complexity and consistently observed higher expression yields compared to four commercial codon optimization tools.
The related research findings, titled "Pichia-CLM: A language model–based codon optimization pipeline for Komagataella phaffii", have been published in PNAS.
Research highlights:
* Pichia-CLM uses host genome data to learn the amino acid-to-codon mapping unbiasedly, taking into account not only host preferences but also position dependence and long-range contextual relationships.
* Pichia-CLM was experimentally validated on six proteins of varying complexity, consistently showing higher expression yields.
* The amino acid and codon embeddings learned by the model can be grouped according to their physicochemical properties, indicating that the language model can capture physically meaningful patterns.
Paper address:
https://www.pnas.org/doi/10.1073/pnas.2522052123
Follow our official WeChat account and reply "Pichia pastoris" in the background to get the full PDF.
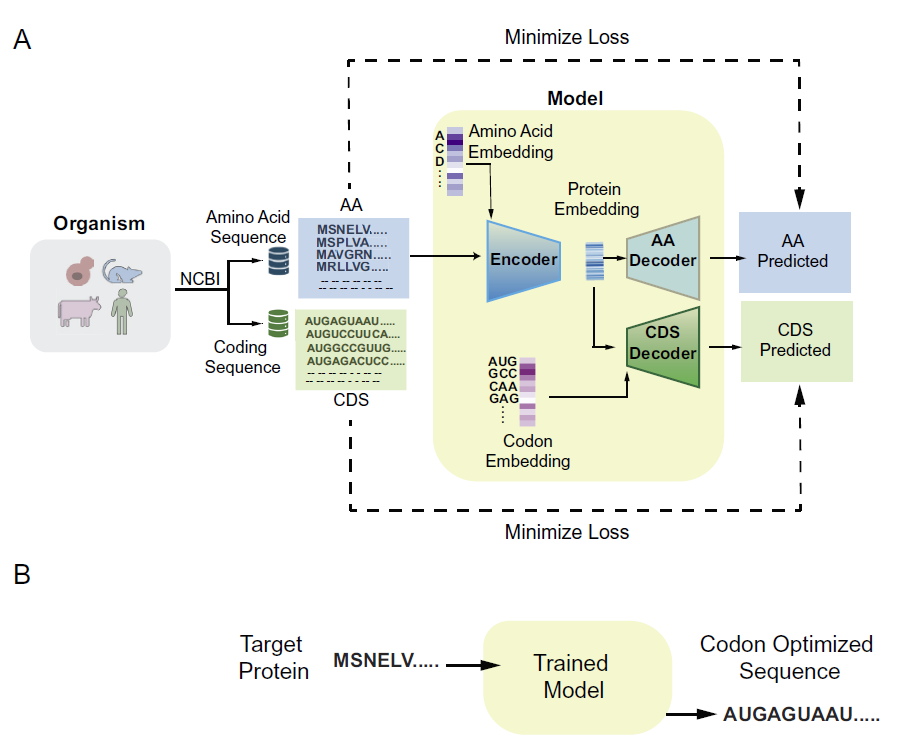
Construct a large-scale sequence dataset centered on Pichia pastoris
Unlike traditional methods that rely on empirical rules, the core idea of Pichia-CLM is to learn coding patterns directly from the host genome. To this end,The research team constructed a large-scale sequence dataset centered on Pichia pastoris.
To train Pichia-CLM, researchers collected amino acid and coding sequence data for two Pichia pastoris variants from NCBI: CBS7435 and GS115. This was supplemented by data previously completed in their laboratory, including genome sequencing and annotation of GS115, K. phaffii (NRRL Y11430), and K. pastoris.In total, approximately 27,000 pairs of amino acid-coding sequence data were used.
During data processing, researchers tokenized amino acids and codons, and introduced starters (…). ),termination( ) and fill ( The dataset is labeled to enable the model to handle sequences of varying lengths and support batch training. Additionally, the dataset is divided into training and test sets, with approximately 201 TP3T used to evaluate the model's predictive ability on unseen data.
It is worth noting that this data construction method does not introduce any artificial "optimization objectives," but is entirely based on natural genome data. This means that the model learns the host's true expression preferences, rather than artificially set approximate rules, laying the foundation for subsequent performance improvements.
Pichia-CLM employs a GRU-based encoder-decoder architecture.
Model Architecture
The Pichia-CLM employs an encoder-decoder architecture based on gated recurrent units (GRUs).GRU is an improved recurrent neural network architecture designed to capture long-range and short-range dependencies in sequence data. By regulating information flow through gating mechanisms, GRU effectively alleviates the vanishing gradient problem common in traditional RNNs. Furthermore, GRU's performance is comparable to Long Short-Term Memory (LSTM) networks, but it requires fewer parameters and consumes fewer computational resources, thus offering greater efficiency advantages in many sequence modeling tasks.
Compared to another mainstream architecture, Transformer, GRU has higher computational efficiency and lower resource consumption on small to medium-sized datasets.Studies have shown that with a data size of approximately 27,000 sequences, introducing a Transformer can actually increase unnecessary complexity, while GRU can achieve a better balance between performance and efficiency.
The model takes the amino acid sequence of a protein as input and generates the corresponding DNA sequence based on patterns learned from the host amino acid sequence and coding sequence. The overall architecture is shown in the figure below:
Model training process
During training, the researchers used a validation set (20% of the training set) for early stopping to optimize the parameters. Simultaneously, hyperparameter selection was performed with the goal of minimizing the validation set loss (sparse classification cross-entropy).The hyperparameter optimization employs a global optimization strategy called Bayesian optimization, combined with code implemented internally by the researchers.
Specifically, the model involves the following hyperparameters:
* Amino acid embedding dimension
* Codon embedding dimension
* Number of units in the encoder layer
* Size of the codon fully connected layer in the decoder
* Size of the amino acid fully connected layer in the decoder
During model training, the decoder input is the actual encoded sequence (i.e., the real codons). In the prediction phase, the model uses the codon predicted at the previous position as the input for the next position, thus achieving fully autoregressive prediction. Sequence prediction terminates when a stop codon is encountered.
After completing the architecture selection and validating the predictive power on the test set, the researchers retrained the final model using the full dataset and continued to employ an early stopping strategy to avoid overfitting. This final model was used to design the coding sequences of exogenous proteins.
Pichia-CLM can generate high-protein-producing constructs.
In the experimental validation section, the research team selected six proteins with different levels of complexity for testing, including:
Human growth hormone (hGH)
* Human granulocyte colony-stimulating factor (hGCSF)
* VHH Nanobody 3B2 (34)
* Engineered SARS-CoV-2 RBD subunit variant (RBD) (35)
* Human serum albumin (HSA)
* IgG1 monoclonal antibody trastuzumab (Trast)
Performance of Pichia-CLM in enhancing protein secretion in Pichia pastoris
first,Researchers selected three human-derived proteins of varying sizes and complexities: hGH, hGCSF, and HSA, and compared the differences in protein secretion yield (titer) between gene constructs generated using Pichia-CLM and their native coding sequences.Overall, for proteins such as hGH and hGCSF, the yield increase was approximately 25%; while for HSA, a significant increase of approximately 3-fold was observed.
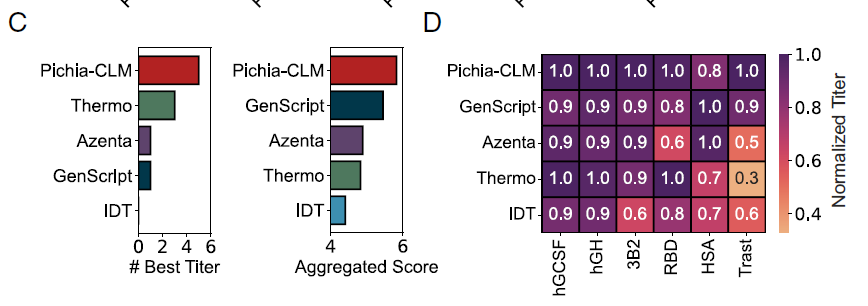
Subsequently, the researchers compared Pichia-CLM with four commercial codon optimization tools: Azenta, IDT, GenScript, and Thermo Fisher (Thermo), evaluating six proteins using two metrics:
* BestTiter: The number of proteins with the highest titer obtained by a particular method.
* Aggregated Score: The sum of the relative titers of different proteins (normalized to the maximum value).
Overall,Pichia-CLM outperformed commercial algorithms in both metrics (Figure C below); it achieved the highest titer in 5 out of 6 proteins, with only a slight decrease in overall score (approximately 0.2) in HSA due to a slightly lower titer (Figure D below).
Assessment of genetic sequence characteristics
After validating the performance of Pichia-CLM in the production of exogenous proteins, the researchers further analyzed the genetic sequence characteristics of different designed constructs.Including other reported protein language models, codon optimization typically relies on one or more codon usage bias (CUB) metrics for design or evaluation. Therefore, this study used data from six test proteins to evaluate the correlation between these CUB metrics and protein yield.
The results showed that none of these indicators exhibited a consistent and high correlation with yield across different proteins. For example, in the case of HSA (as shown in Figure A below), the maximum positive correlation with codon volatility and codon frequency distribution (CFD) was only 0.43, while the maximum negative correlation with codon pair score (CPS) was only 0.25.
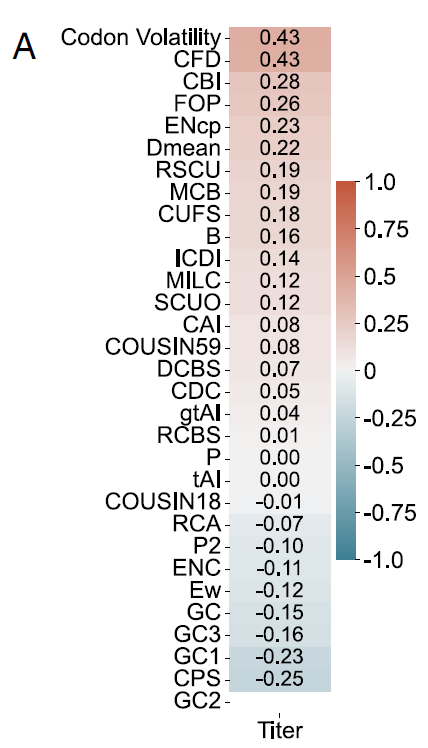
Global CUB metrics calculated based on the entire sequence have significant limitations in characterizing features related to exogenous protein production.This further demonstrates the need for new evaluation metrics to assess codon optimization tools, combined with rigorous experimental validation of diverse proteins—a result that directly challenges the theoretical foundation of traditional codon optimization.
Sequence feature evaluation
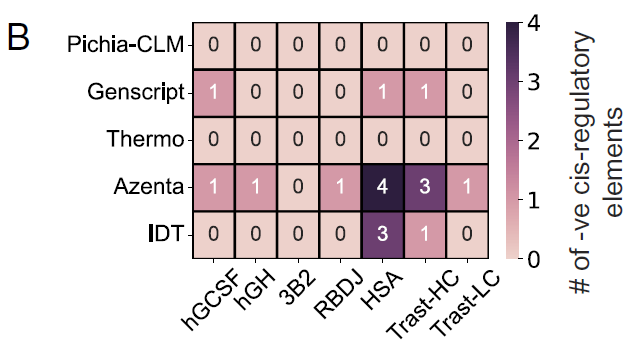
The researchers also assessed the presence of negative cis-regulatory elements in different codon-optimized constructs, which could interfere with the host's regulatory mechanisms and should therefore be avoided as much as possible in exogenous DNA sequences.
Of the 6 tested proteins,No negative cis-regulatory elements were detected in constructs designed using Pichia-CLM; in contrast, GenScript contained one negative cis-regulatory element in three out of six proteins; Azenta and IDT generated sequences containing three to four such elements in at least one protein.As shown in Figure B:
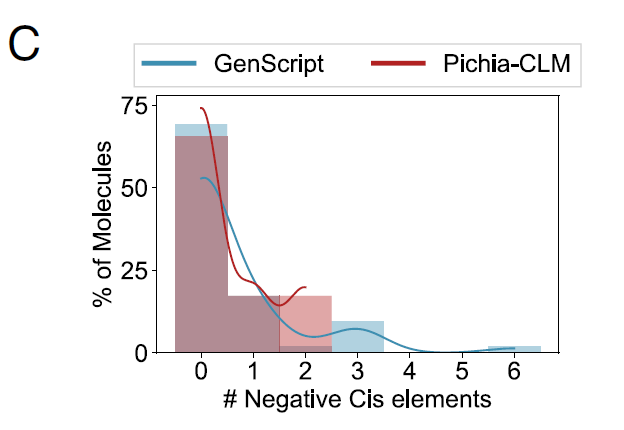
Researchers also analyzed the performance of Pichia-CLM in 52 biotechnology-related proteins, and the results showed:The protein sequence of 75% contains no negative cis-regulatory elements at all, while the remaining 25% contains at most two such elements.In contrast, the best-performing commercial algorithm, GenScript, still produced constructs containing 3 to 6 negative cis-regulatory elements in proteins of approximately 15%, as shown in Figure C below:
In summary, these results demonstrate that Pichia-CLM can not only generate high-yield protein constructs, but also learn key genetic sequence features and achieve a balance among multiple factors, thereby designing robust coding sequences suitable for host expression.
AI accelerates the industrialization of protein production
In the biopharmaceutical industry, improving protein production efficiency has always been a key factor determining the success or failure of research and development translation and commercialization. From monoclonal antibodies to recombinant vaccines, and then to various fusion proteins and enzyme preparations, market demand continues to grow, and the requirements for yield, stability, and consistency are constantly increasing.
To achieve this goal, the industry has developed a multi-layered optimization system: at the host level, in addition to traditional E. coli and Saccharomyces cerevisiae, Pichia pastoris and mammalian cells have become the mainstream production platforms due to their superior post-translational modification capabilities and expression efficiency; at the molecular design level, in addition to codon optimization, this includes promoter strength regulation, signal peptide screening, mRNA structural engineering, and protein folding and secretion pathway optimization; and at the process level, high-density fermentation, feed strategy optimization, and bioreactor parameter control also play a decisive role in the final yield.
Outside of this system,A new type of "decellularization" technology is rapidly emerging: Cell-Free Protein Synthesis (CFPS).This technology bypasses the cell growth process and directly utilizes the transcription and translation system in cell lysates to achieve rapid protein expression. It has been widely used in the development and production of antibodies, enzymes, and even antibody-drug conjugates. However, the CFPS system itself is a highly complex multivariate system involving dozens of components such as DNA templates, enzyme systems, energy donors, amino acids, and ionic environments. Its combination space is extremely large, and traditional experience-based optimization methods often fail to achieve an ideal balance between cost and yield.
Against this backdrop, AI-driven automated optimization is demonstrating disruptive potential. Recently, OpenAI, in collaboration with leading synthetic biology company Ginkgo Bioworks, released groundbreaking research findings.The "closed-loop automation system" built on the GPT-5 large language model has successfully achieved dual optimization of cell-free protein synthesis (CFPS) technology—reducing the total production cost of the technology by 401 TP3T, significantly reducing reagent costs by 571 TP3T, and improving protein synthesis titer by 271 TP3T.
In the future, similar approaches will be extended to a wider range of biomanufacturing scenarios. From optimizing metabolic pathways in cell factories to real-time control of fermentation processes and intelligent design of expression constructs, artificial intelligence is gradually being embedded in all aspects of protein drug production.
References:
1.https://www.pnas.org/doi/10.1073/pnas.2522052123
2.https://phys.org/news/2026-02-ai-yeast-dna-language-boost.html#google_vignette
3.https://mp.weixin.qq.com/s/Qkl6j9HcFB7W_Y5Xh-9BCw