Command Palette
Search for a command to run...
A Team From the Chinese University of Hong Kong, Zhejiang University, and Macao Polytechnic University Proposed a General Framework, Bi-TEAM, to Improve the Accuracy of Hemolytic Disease Prediction by 350%, Integrating Biological Semantics and Chemical precision.

In the fields of biochemistry and molecular engineering, characterization learning is gradually becoming a key technology for elucidating molecular functions and advancing the discovery of therapeutic molecules. The quality of embedded features often determines the performance ceiling of downstream tasks such as peptide property prediction and de novo design. As a core molecule connecting biological function and chemical properties, peptide structure and function modeling has significant value in drug development.In recent years, the introduction of non-classical amino acids has significantly expanded the functional space of peptides and improved their stability and bioavailability, but complex chemical modifications have also brought new challenges to traditional modeling methods.How to integrate biological evolutionary information and chemical rationality into a model simultaneously is becoming a key issue that urgently needs to be addressed in this field.
Currently, peptide modeling is mainly carried out along two technical paths.on the one hand,Protein language models, such as ESM and ProtT5, capture biological context and evolutionary information through large-scale sequence pre-training, providing transferable biological representations for downstream tasks.on the other hand,To address the issue of modifications to non-classical amino acids, researchers used a chemical language model to capture chemical details through atomic-level word segmentation, thus compensating for the shortcomings of protein models at the chemical level.
However, both types of models have inherent limitations. Protein language models are limited by the natural amino acid character set, making it difficult to handle non-classical residues. Existing methods for approximating or expanding vocabulary often introduce bias or lead to semantic sparsity. Chemical language models, on the other hand, ignore the global biological context, and dense word segmentation easily exceeds the context window, making it difficult to adapt to long sequence modeling. General models also suffer from domain bias.
To address the aforementioned issues, the Chinese University of Hong Kong, in collaboration with the Macao Polytechnic University, Zhejiang University, Xiangya Second Hospital of Central South University, and the University of Electronic Science and Technology of China, proposed a selective fusion modeling paradigm.Based on the understanding that "chemical variation is a local perturbation of the biological semantic space", a general framework Bi-TEAM was designed to inject local chemical variation into the global protein background.
This framework uses biological representations as its semantic backbone and achieves an effective fusion of biological evolutionary information and chemical rationale through adaptive injection of chemical signals. In multiple tasks, Bi-TEAM consistently outperforms state-of-the-art baseline models: under rigorous data partitioning based on skeletal similarity, the Matthews correlation coefficient is improved by up to 661 TP3T; in the hemolysis prediction task, accuracy is improved by 3501 TP3T.
The related research findings, titled "Bi-TEAM: A Unified Cross-Scale Representation Learning Framework for Chemically Modified Biomolecules," have been published as a preprint on arXiv.
Research highlights:
The Bi-TEAM framework can adaptively integrate multi-scale biochemical properties and serve as a high-fidelity prior model for efficient peptide design.
* Researchers comprehensively evaluated Bi-TEAM on 10 diverse datasets across 3 biochemical domains, achieving state-of-the-art (SOTA) performance in 7 key prediction tasks.
* This model achieves a dual breakthrough in prediction and generation tasks, improving MCC by 66% under strict scaffold similarity segmentation, while simultaneously increasing the success rate of designing cell-penetrating cyclic peptides by nearly 4 times.

Paper address:
https://arxiv.org/abs/2603.01873
Follow our official WeChat account and reply "Bi-TEAM" in the background to get the full PDF.
A comprehensive evaluation was conducted covering three major biochemical fields and 10 diverse datasets.
This study evaluates the properties from two dimensions: property prediction and guided generation, covering three major research areas: modified peptides, post-translational modifications (PTMs), and natural proteins, involving a total of 10 datasets.
In the field of modified peptides, research focuses on evaluating the model's ability to predict membrane permeability.The core training data comes from the ProPAMPA database, which contains 12–46 ring atoms, with a sequence length distribution that is approximately normal, but exhibits significant long tails at both ends. It also contains a large number of natural and non-classical amino acid residues, demonstrating high chemical diversity. After deduplication using RDKit,It contains a total of 6,876 non-conjugated cyclic peptide sequences.
To evaluate the model's generalization ability, the study further introduced three external wet experimental datasets: ProCacoPAMPA, CycPeptMPDB v1.2, and the Rezai dataset. These datasets cover cyclic peptide samples of different lengths and structural types. Specifically:
ProCacoPAMPA:All transmembrane cyclic peptide sequences of lengths 6 and 10 were collected from existing studies and constructed into a standardized dataset.
CycPeptMPDB v1.2:The latest version of the largest publicly available non-classical cyclic peptide membrane permeability database, compiled from 56 papers, contains 8,466 records. In this study, researchers removed samples that were duplicates of the ProPAMPA dataset, ultimately obtaining a refined subset containing 1,230 data points.
Rezai:This includes passive membrane permeability data for 11 cyclic peptides. These data were obtained through PAMPA experiments and are often used for external model validation under small sample conditions.
To further validate the drug-likeness and disease association of the model,Researchers conducted a drug-likeness prediction task on the PTM dataset.The data used included two categories: drug-grade datasets and disease-associated datasets. The former mainly consisted of longer protein sequences, with modification sites exhibiting a distinct long-tail distribution. The latter primarily came from databases such as dbPTM and genome-wide association studies (GWAS), and while the distribution of modification sites was similar to that of the former, the sequence length range was wider, thus providing a more diverse structural context.
In the field of natural proteins, researchers focused on evaluating the model's performance in solubility and hemolysis prediction tasks to explore the key mechanisms of peptide hemolysis and changes in protein solubility. The datasets used primarily included three categories: hemolysis, anticontamination, and solubility. Among them:
Hemolysis data are from the DBAASP v3 database.It contains a total of 9,316 sequences composed of classic L-type amino acids.
The anti-pollution dataset mainly consists of short peptide sequences.The lengths are concentrated between 5 and 10 amino acid residues, and their LogP distribution is approximately normal. The samples exhibit a good clustering structure in the feature space.
The solubility dataset is derived from protein sequences annotated with PROSO II.Its label is based on a retrospective analysis of the Protein Structure Initiative.
Bi-TEAM: A unified cross-scale characterization learning framework for chemically modified biomolecules
Bi-TEAM aims to address the challenge of existing single-modal models in simultaneously capturing global evolutionary biological information and local chemical structural details (fine-grained chemical space). As shown in the figure below,Its core idea is to construct a dual-perspective representation system that deeply integrates evolutionary biological space and chemical structure space.This provides more accurate modeling capabilities for peptide sequences containing non-classical amino acids.
In terms of overall architecture,The model uses the biological space constructed by the protein language model as its semantic backbone, making full use of the evolutionary patterns and contextual relationships learned from large-scale natural sequences.Meanwhile, a chemical language model (CLM) is introduced to capture structural information at the atomic level, compensating for the inherent limitations of the protein language model (PLM) in handling chemical modifications. The two types of models complement each other at the representation level, jointly expanding the expressive power of the input sequence.

When processing modified peptide sequences, Bi-TEAM encodes them using two complementary information streams:One is a biological sequence stream.Modified amino acids are mapped to the structurally closest natural amino acids, thereby avoiding word segmentation table expansion and preserving evolutionary semantics that can be used for modeling.The other is the SELFIES-like representation stream.It is used to accurately describe the functional group changes and chemical bond structures of modified residues at the atomic level, providing stable structural information for chemical language models.
After completing dual-stream encoding,The model is fused using a dual-gated residual mechanism guided by position-aware embellishment cues:Using biological representations as the semantic backbone, key chemical signals are filtered and injected using gating units, while residual connections of biological features are preserved. This allows the model to establish an effective correlation between global sequence constraints and local chemical changes while maintaining training stability.
At the application level, Bi-TEAM has good versatility.When processing unmodified natural protein sequences, the model can directly omit the mapping and localization steps, adapting to routine protein tasks without adjusting the overall architecture.
In terms of training strategies,The study employs a two-stage framework of "pre-training-fine-tuning":First, domain-adaptive pre-training was performed on two types of basic encoders on natural protein sequences and small molecule chemical corpora, respectively. Then, through multi-task joint fine-tuning, the model learned the fusion rules of biological and chemical features in different task scenarios, thereby further improving the overall generalization ability.
Bi-TEAM has achieved a breakthrough in the design of penetrating cyclic peptides, increasing the success rate by 4.6 times.
To verify the application capabilities of Bi-TEAM in unknown chemical spaces, this study focuses on non-canonical cyclic peptide design for cell-penetrating treatment of neovascular age-related macular degeneration (nAMD) using non-invasive drug delivery as a scenario. The study systematically carried out a full-process experiment of "prediction-guided analysis" to evaluate the performance of the model in property-guided molecular design.
nAMD is a major cause of irreversible blindness in the elderly, and its core pathology is VEGF-driven choroidal neovascularization and leakage. Currently, clinical treatment mainly relies on intravitreal injection of large-molecule anti-VEGF drugs (such as Aflibercept, 115 kD).However, these drugs have difficulty penetrating the physiological barrier of the eye, and long-term injections can lead to complications and compliance issues.Designing peptide binders that can specifically bind to Aflibercept and promote its transbarrier transport would offer new possibilities for non-invasive eye drop therapy. Compared to easily degradable linear peptides with short half-lives, cyclic peptides, with their more stable structure and higher permeability, are considered more ideal delivery carriers, which is the main motivation for researchers to focus on cyclic peptide design.
This study first conducted a prediction and assessment of cell-penetrating peptides (CPPs) to provide a foundation for subsequent generation tasks.The dataset was constructed according to the pLM4CPP standard scheme, integrating databases such as CPPsite2.0, C2Pred, and CellPPD. After screening and deduplication, 1,399 positive samples (experimentally validated penetrating peptides) and 4,080 negative samples were obtained. Comparison models included mainstream protein embedding models such as SeqVec, ESM2, and ProtT5, and evaluation metrics covered ACC, BACC, Sn, Sp, MCC, and AUC.
The results show that Bi-TEAM achieved the best performance across all metrics:The ACC was improved by 5.521 TP3T compared to SeqVec, the BACC by 5.881 TP3T compared to ESM2-480, the Sn by 12.581 TP3T, the Sp by 1.451 TP3T compared to ProtT5-XL BFD, the MCC by 14.681 TP3T compared to SeqVec, and the AUC by 8.451 TP3T compared to ESM2-480. The significant improvements in sensitivity and MCC indicate that the model has a clear advantage in identifying true penetrating peptides.
Building on this foundation, further research will be conducted on property-guided cyclic peptide generation experiments.Using BoltzDesign1 as the baseline framework, 1,000 cyclic peptides of length 10-20 were generated under two conditions: one using only the default structural constraints, and the other introducing Bi-TEAM as an additional gradient guide during the generation process.
The success criterion was a Bi-TEAM prediction log odds greater than 0.5. The results showed that...Traditional methods yielded a success rate of only 6.71 TP3T in generating cell-penetrating cyclic peptides, while Bi-TEAM-guided methods improved this to 30.71 TP3T.Meanwhile, the structural quality did not decline: the average pLDDT of the generated peptide-Aflibercept complex exceeded 0.82, indicating that the model maintained good structural confidence and binding interface stability while improving penetration.

To understand the guidance mechanism, researchers further analyzed the residue patterns of the generated sequence. Previous studies have shown that the hydrophobic triplet composed of tryptophan (W), phenylalanine (F), and tyrosine (Y), as well as positively charged residues such as arginine (R) and lysine (K), are key features for cell-penetrating peptides to achieve membrane transport.
Analysis revealed that,Guided by Bi-TEAM, the co-occurrence frequency of hydrophobic triplet and two positively charged residues in the generated sequence was significantly increased, and the residue number distribution also showed a consistent trend.This enrichment pattern is highly consistent with the known structure-function rules of penetrating peptides, indicating that Bi-TEAM can not only capture relevant biological mechanisms, but also significantly increase the probability of sequences with membrane-penetrating properties during the generation process. Control variable analysis further ruled out the influence of peptide length (10–20 residues), showing that the model indeed guides the sampling distribution to a chemo-biological co-location space more favorable for membrane transport.
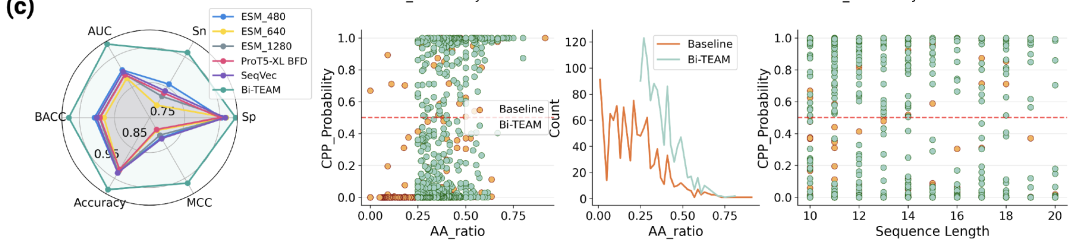
In the middle section: the relationship between the abundance of key hydrophobic residues and the penetration probability;
Right: Relationship between cyclic peptide length and penetration probability
Finally, the study validated the results at the structural level through a case study. Researchers first visualized the three-dimensional structure of the Aflibercept dimer and colored its molecular surface based on electrostatic potential; subsequently, they used AlphaFold3 to predict and design the complex structure of the cyclic peptide with Aflibercept. Analysis identified two potential cyclic peptide binding pockets: one a hydrophobic cavity composed of three rings, and the other formed by a ring structure and a β-sheet fragment. This structural information provides important basis for subsequent cyclic peptide optimization and potential clinical applications.

Focusing on technological innovation in the field of peptide drug development
In the field of peptide science, from basic research to clinical translation, a host of research institutions around the world are actively exploring new technological pathways and treatment options to conquer major diseases.
For example, the structural biology team at the School of Biochemistry, University of Bristol, UK.Advanced techniques such as cryo-electron microscopy and X-ray crystallography are used to analyze the fine structure of the immune system, and on this basis, structure-guided peptide drug design is carried out.They are attempting to develop next-generation drug candidates for the treatment of autoimmune diseases by designing cyclic peptide molecules that can precisely activate the human complement system.
Meanwhile, the ToxiCode project, a collaboration between King's College London and the University of Zagreb, explores a unique path to discovering new drugs from animal venom.The project combines artificial intelligence and synthetic biology, using a hybrid AI system to learn peptide sequence patterns and their structure-activity relationships, enabling the rapid design of novel bioactive peptides targeting cancer, neurological disorders, and infectious diseases.This provides a new methodological framework for sustainable and ethical drug discovery.
This demonstrates that peptide drug development is gradually forming a new research paradigm: structural biology, artificial intelligence, and chemical biology are increasingly converging, and the boundaries between basic research and industrial development are becoming increasingly blurred. New molecules often emerge from interdisciplinary technological combinations, but what truly determines whether they can reach clinical application is the gradually established translational pathway from laboratory discovery to industrialization. In this process, peptide molecules, due to their unique properties between small and large molecules, are being re-evaluated and are showing new applications in an increasing number of disease areas.
Reference Links:
1.https://www.bristol.ac.uk/news/2025/november/bristol-researcher-awarded-over-850000-to-develop-new-treatments.html
2.https://www.kcl.ac.uk/news/kings-to-collaborate-in-venom-based-drug-discovery-project
3.https://mp.weixin.qq.com/s/X67D1qrUzclwOsJ9cKUtZg








