Command Palette
Search for a command to run...
RNA Structure Prediction Rivals AlphaFold 3! A Virginia Tech Team Proposes RNAbpFlow, Which Is Completely Independent of Evolutionary information.

In November 2020, AlphaFold 2 rose to fame at the CASP14 international protein structure prediction competition. With its near-experimental accuracy in predicting the three-dimensional structure of proteins, AlphaFold 2 not only solved the protein folding problem that had plagued the biological community for half a century, but also propelled its two creators to the Nobel Prize in Chemistry four years later with its stunning results. However, the success of AlphaFold 2 was just the beginning. Like a nuclear bomb, it ignited the entire field of structural biology, sparking a wave of research using artificial intelligence to analyze the spatial conformation of biological macromolecules.
Inspired by AlphaFold 2's groundbreaking achievement in the field of proteins, high hopes are also placed on using artificial intelligence to solve the problem of predicting the three-dimensional structure of ribonucleic acid (RNA).However, current mainstream algorithms still face several unavoidable practical bottlenecks when they are implemented:First, most Transformer-based RNA prediction models heavily rely on dominant evolutionary sequence information provided by multiple sequence alignments (MSA) or homologous sequence information indirectly obtained from biological language models. However, the isostructural nature of RNA base pairing makes it difficult to generate high-quality, reliable MSA results. Second, most mainstream methods fail to fully explore the key information of base pairing in RNA secondary structure, which is the core factor determining the three-dimensional folding morphology of RNA. Third, RNA molecules possess natural conformational flexibility and exist in multiple stable spatial conformations. Existing algorithms mostly only output single static results and cannot reconstruct the true dynamic conformation set of RNA.
To address the aforementioned challenges, Professor Debswapna Bhattacharya and his student Sumit Tarafder of Virginia Tech collaborated to develop an SE(3)-equivariant flow-matching model, RNAbpFlow, based on sequence and base pair conditions.This model can generate a complete set of all-atom RNA conformations solely based on RNA nucleotide sequences and base pairing information, without requiring evolutionary information such as multiple sequence alignments or homologous templates.It addresses several shortcomings of existing AI-based RNA structure prediction methods. Multiple benchmark experiments have demonstrated that introducing base pairing as a constraint can significantly improve the model's prediction accuracy. In the two core tasks of RNA topology sampling and 3D conformation modeling, RNAbpFlow outperforms existing mainstream methods.
It is worth mentioning that in the comparative test between RNAbpFlow and AlphaFold 3, a set of impressive data revealed the final outcome of the game:In the CASP16 blind test experiment, RNAbpFlow was able to accurately reconstruct the global folding topology of the vast majority of the 14 RNA targets (≤200 nt), with 12 of them meeting the qualified prediction criteria.In contrast, under the same experimental conditions, only 8 target predicted structures of AlphaFold 3 met the standard for matching with the natural conformation.
The relevant research results, titled "RNAbpFlow: base pair-augmented SE(3) flow matching for conditional RNA 3D structure generation", have been published in Nature Methods.
Research highlights:
- The proposed RNAbpFlow is an SE(3) isofluidic matching model based on sequence and base pair conditions, which addresses several shortcomings of existing RNA three-dimensional structure prediction methods.
- Three major innovations were proposed: three-channel base pairing conditional input, nucleoside base center characterization, and base pairing-specific auxiliary losses (bp2D and bp3D), which significantly improved the fidelity of base pairing and three-dimensional structure.
- We propose an independent and non-repetitive training and testing set strategy, and use multiple authoritative benchmarks and CASP blind testing for validation. The overall performance outperforms existing mainstream algorithms.

Paper address:
https://www.nature.com/articles/s41592-026-03128-4
Independent deduplication of training and test sets ensures true and fair evaluation.
To build the self-developed model RNAbpFlow and ensure the authenticity and reliability of its evaluation results, this study uses independent training and test sets with no content duplication, and assigns specific model weights to different test benchmarks, rather than using a single default model throughout the process, in order to avoid training data being mixed into the evaluation process and causing result distortion.
Regarding model development and internal validation,This study uses the RNA3DB dataset. This dataset is highly suitable for training deep learning models and internal benchmarking, possessing non-redundancy at both the sequence and structural levels. In this study, the dataset used was the RNA3DB dataset version parsed from the Protein Database (PDB) on April 26, 2024, and the training-test partitioning scheme given in the original paper was followed to select representative RNA sequences for the experiments.
To ensure the dataset contains only high-quality natural RNA structures and accurate base pairing information, the experiment underwent multi-layered quality control filtering. This included removing structures with only a single atom in a single nucleotide and structures mixed with protein residues; truncating continuous experimental sequences to correct mismatches between FASTA sequences and three-dimensional structures, protecting the integrity of base pairings; and deleting sequences without base pairings from natural structures. After extracting base pairing information using RNAView, filtering out continuous unpaired RNA chains with a length ≥ 20 nucleotides, and limiting the sequence length to 30-200 nucleotides, a pure training set containing 560 RNA sequences (sequences that cannot match the Rfam family were removed to further reduce the risk of data leakage) and a pure test set containing 48 sequences were finally obtained.
It is worth mentioning that this part of the data partitioning fully follows the non-redundant splitting of RNA3DB, and there is no overlap in sequences or structures between the training and test sets, so as to avoid data leakage from the root.
In the comparative experiment of RNAbpFlow and RNAJP,The study used a dataset of 22 RNA sequences containing three-way connectors, similar to those used in the RNAJP study, and then performed a screening process. Researchers first removed multimer structures from this dataset and then quickly compared the remaining sequences with the aforementioned clean training set to remove redundancy and eliminate sequence duplication. The final processed dataset was reduced to 12 sequences.
Regarding compatibility with CASP15 and CASP16 international competitions,In order to strictly adhere to the blind testing rules of the competition and ensure a fair comparison with other algorithms, the study did not directly adopt the inherent training and test set partitioning provided by RNA3DB. Instead, it reorganized and planned two completely disjoint and non-overlapping new training sets from RNA3DB.
For CASP15, since the first batch of RNAs to be tested in the CASP15 competition was only released to the public in May 2022, researchers only included RNA sequences uploaded to the PDB database before April 2022 (all from RNA3DB) when designing the new training set. This ensured that the model training completely avoided the RNAs to be tested in CASP15, strictly adhering to the blind testing rules. Ultimately, the training set yielded 731 RNA sequences, ranging in length from 30 to 784 nucleotides. The test set used for model evaluation consisted of 6 natural RNAs and 4 synthetic RNAs from the CASP15 benchmark set.
For CASP16,The training set used in the experiment consisted of 994 experimentally resolved PDB structures and 2,170 high-confidence prediction structures used for cross-distillation data augmentation, for a total of 3,164 samples.The ratio of PDB structure to cross-distilled data in each training batch is approximately 1:2.2.
Specifically, for the same reason, the first batch of RNAs to be tested in CASP16 was released in May 2024. Therefore, researchers merged all eligible data from RNA3DB into a single training set, including only PDB structures uploaded on or before April 6, 2024. After rigorous screening, 994 RNA sequences and their corresponding measured three-dimensional structures were obtained. The test data came from 28 RNAs with actual experimental three-dimensional structures currently available in CASP16.
The data used to construct the cross-distillation dataset came from the bpRNA-1m(90) dataset, primarily to explore the effects of data augmentation. The bpRNA-1m(90) dataset contains 28,370 RNA sequences (and their corresponding secondary structures). After redundancy removal and limiting the sequence length to 30-200, and after multiple extractions using the MMseqs2 clustering tool,The final result was a cross-distillation dataset of 2,170 high-confidence RNA structures.
Conditional generation framework based on stream matching
RNAbpFlow is an SE(3) isofluidic matching model based on sequence and base pair conditions (as shown in the figure below). It is modified from the protein structure generation model FrameFlow and adopts the nucleotide representation method proposed by NuFold, representing each nucleotide in the RNA sequence as a rigid frame. Using nucleotide sequence and base pairing information as constraints, it predicts dihedral angles through iterative sampling, gradually reducing all atoms to their final, complete three-dimensional structure of RNA with the true ribose fold conformation. In short,The core advantages of RNAbpFlow can be summarized in two points: "input conditions" and "model paradigm".
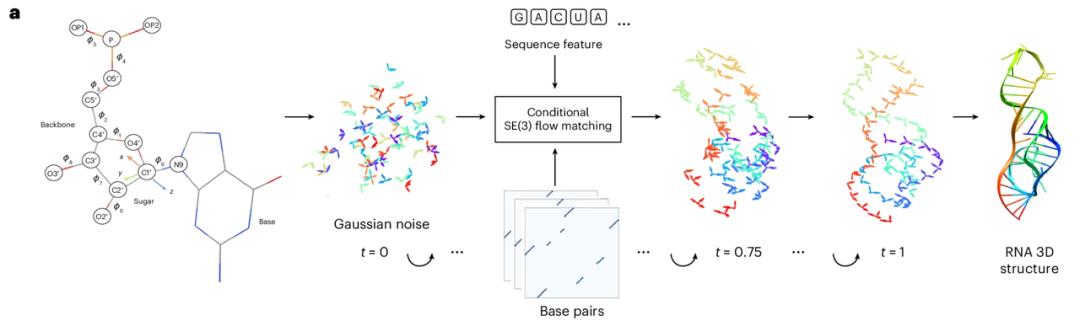
Regarding the "input conditions",RNAbpFlow can conditionally generate three-dimensional RNA structures without any MSA or homologous template information, relying solely on sequence and base pairing information.
First, for the sequence, an L-length RNA sequence is input, and nucleotides are represented using one-hot encoding, i.e., a binary vector containing four elements corresponds to the four types of nucleotides (A, U, C, and G). Second, for the base pairing information processing during the training phase (this is the key innovation), three tools—RNAView, MC-Annotate, and DSSR—are used to extract two-dimensional pairing annotations from the experimentally resolved natural three-dimensional structure, and these are represented as three independent L × L binary matrices. To fully capture various classical and non-classical base pairing features, the study does not uniformly correct the contradictory annotations obtained by the three tools. Instead, the three binary matrices are directly concatenated into an L × L × 3 tensor, which serves as the input to the bias term in the denoising network structure, providing pairing feature information in three independent channels.
In addition, for the sampling and inference scenarios of the CASP15 and CASP16 competition test targets, since there are no true values for natural base pairings, the experiments used sequence-dependent pairing prediction matrices output by three RNA 2D structure prediction tools: IPKnot, SPOT-RNA, and RibonanzaNet. All three tools support base pairing prediction based on pseudoknot identification, and their selection was based on their sampling performance on natural RNA targets in CASP15. Furthermore, RNAbpFlow is highly versatile, supporting users to define three sets of pairing matrices as input. If only one set of custom pairing matrices is available, it can be copied three times to match the three-channel 2D input format required by the network.
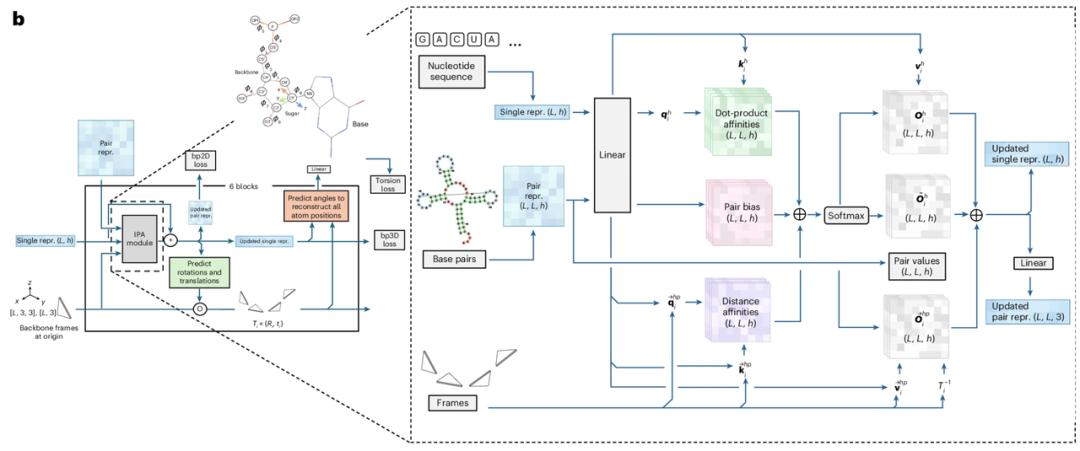
In terms of "model paradigm",The key lies in "flow matching." Flow matching is a type of deep generative model whose core objective is to learn a velocity field (or flow field) that matches the probability flow of the data distribution, thereby transforming simple distributions like Gaussian distributions into the complex data distribution required for the target in a high-dimensional space. Flow matching directly learns this velocity field to describe the motion of sample points migrating from a simple distribution to the target distribution, without completely disrupting the original data distribution. By integrating ordinary differential equations on the learned vector field, flow matching can generate simpler migration trajectories to approximate the target. Compared to diffusion models, flow matching can significantly improve the computational speed of generating large-scale samples.
In this study, the goal of the flow matching method is to learn a parameterized vector field Ut. This vector field represents a smooth, time-varying mapping, from which ordinary differential equations are generated to describe the transformation relationship between two distributions: distribution p₀(T₀) (noisy frames) and distribution p₁(T₁) (true reference frames). To learn this mapping, the researchers trained a parameterized neural network Vθ(Tₜ, t), which predicts the vector field based on the noisy true reference frame Tₜ at time t. This part of the network is designed with reference to FrameFlow, using structural modules from AlphaFold 2 as the backbone network architecture.
Regarding the specific training settings, the experiment was conducted using the PyTorch-Lightning framework to train the model, employing the Adam optimizer with a learning rate of 0.0001. The distributed training process ran on 8 NVIDIA H100 GPUs for 1500 iterations.
Mission-critical performance surpasses AlphaFold 3
To evaluate the sampling performance of RNAbpFlow, the study first compared it with RNAJP, a three-dimensional RNA structure sampling method based on coarse-grained molecular dynamics simulations that explicitly considers base pairing information, covering non-classical base pairing, base stacking interactions, and long-range loop-loop interactions.
The experimental results are shown in the figure below.RNAbpFlow outperformed RNAJP in both evaluation metrics.Specifically, RNAbpFlow achieved a high average local distance difference test score (lDDT score) of 0.66, while RNAJP only achieved 0.59. Similarly, in terms of global topology sampling, RNAbpFlow achieved an average template modeling score (TM-score) of 0.38, while RNAJP achieved 0.32.
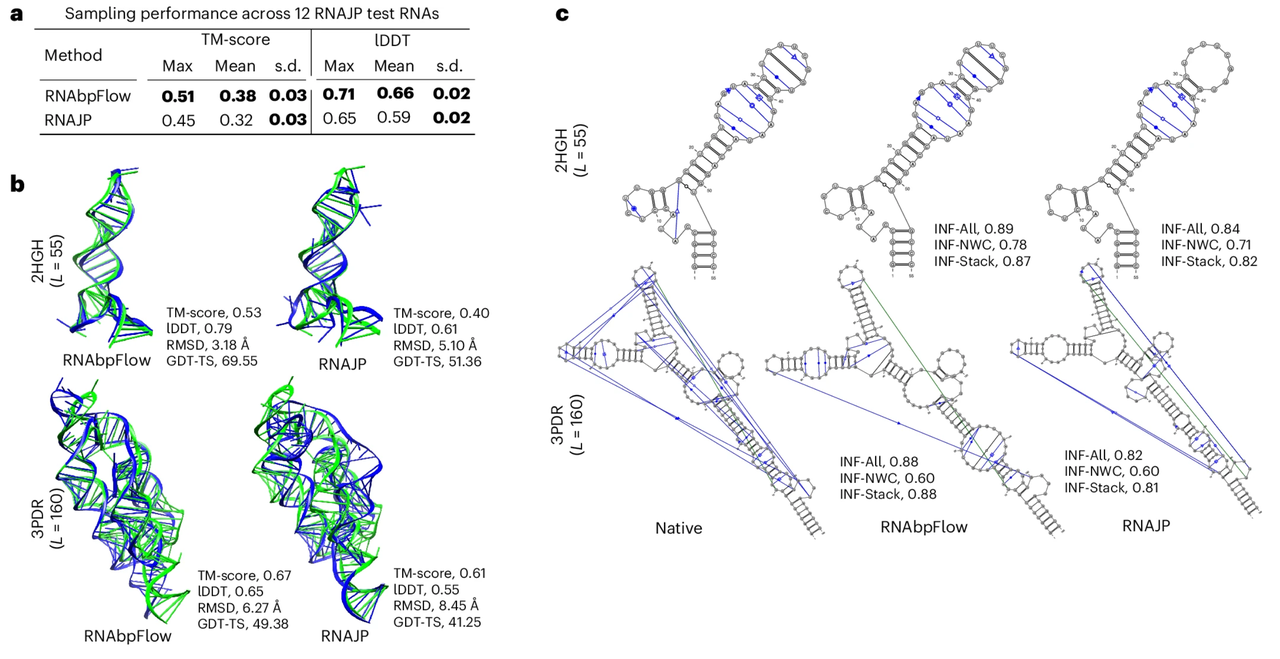
Regarding the evaluation of sampling validity, the correct folding structure is determined based on the TM-score.RNAbpFlow detected RNA targets as high as 66.671 TP3T.The detection rate of correctly folded structures based on lDDT scores was 251 TP3T; in contrast, the rates for RNAJP were only 41.671 TP3T and 0 TP3T, respectively. Furthermore, among all 12,000 simulated conformations generated by RNAbpFlow, 13.41 TP3T conformations had template modeling scores higher than 0.45, and 9.61 TP3T conformations had lDDT scores higher than 0.7. In contrast, among the simulated conformations of RNAJP, only 1.731 TP3T had template modeling scores higher than 0.45, and no conformations had lDDT scores higher than 0.75.
The results above demonstrate that RNAbpFlow not only outperforms RNAJP in scoring optimal structures, but also achieves a higher percentage of high-quality simulated conformations.This highlights its efficiency in dual sampling tasks of global topology and local configuration.
Next, the research team compared RNA bpFlow with several existing methods on the CASP15 blind test dataset (as shown in the table below). The results show that...When real and accurate natural base pairing information is input, the modeling and prediction performance of RNAbpFlow is significantly improved.The average TM-score reached 0.48, the root mean square deviation of all atoms (RMSD) was 7.77, and the non-Watson-Crick base pair interaction network fidelity (INF-NWC) was 0.62; when only the base pairings predicted by the algorithm were used, the three indices were 0.40, 10.70, and 0.48, respectively.
In comparison, when providing true base pairing information, TM-score increased by 201 TP3T, RMSD decreased by 27.41 TP3T, and INF-NWC increased by 29.21 TP3T, highlighting the importance of high-quality base pairing information.
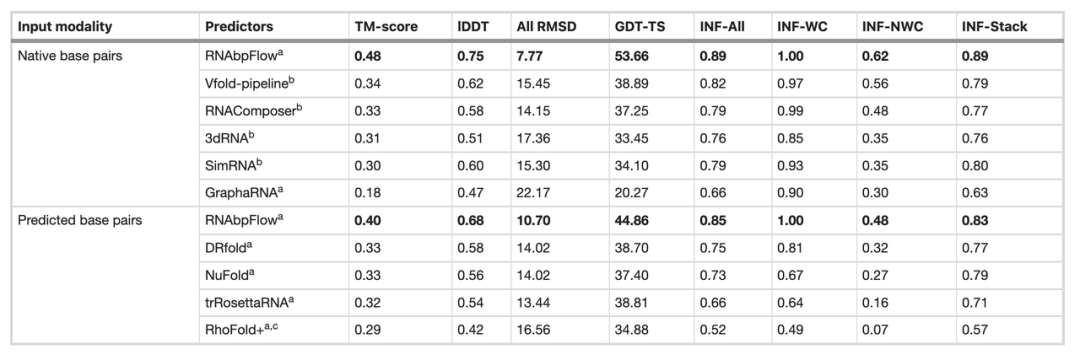
In contrast, other methods show very limited performance improvement even when inputting real natural base pairs. For example, Vfold, the highest-performing method, only achieves a TM-score of 0.34, while RNAComposer has a minimum RMSD of 14.15. This demonstrates that RNAbpFlow possesses greater adaptability, efficiently utilizing precise base pairing constraints in deep generative modeling and significantly raising the upper limit of predictive performance for AI methods.
To verify the sampling accuracy of RNAbpFlow on the CASP16 blind test targets, researchers compared it with the two best-performing prediction models in the CASP16 competition. The results (as shown in the figure below) show that RNAbpFlow outperforms all algorithms, including AlphaFold 3, in terms of average maximum TM-score and lDDT metrics for 14 predicted targets (≤200 nt). Of the structural conformations generated by RNAbpFlow, at least one correctly folded conformation can be found in 12 (85.711 TP3T) of them; in contrast, AlphaFold 3 only achieved this in 8 (57.131 TP3T) conformations, demonstrating the more stable performance of RNAbpFlow.
However, for RNA targets longer than 200 nt, RNAbpFlow still outperforms NuFold, trRosettaRNA2, and DRfold2, but performs worse than AlphaFold3. This is because the fidelity of predicted base pairings for long sequences is significantly reduced.
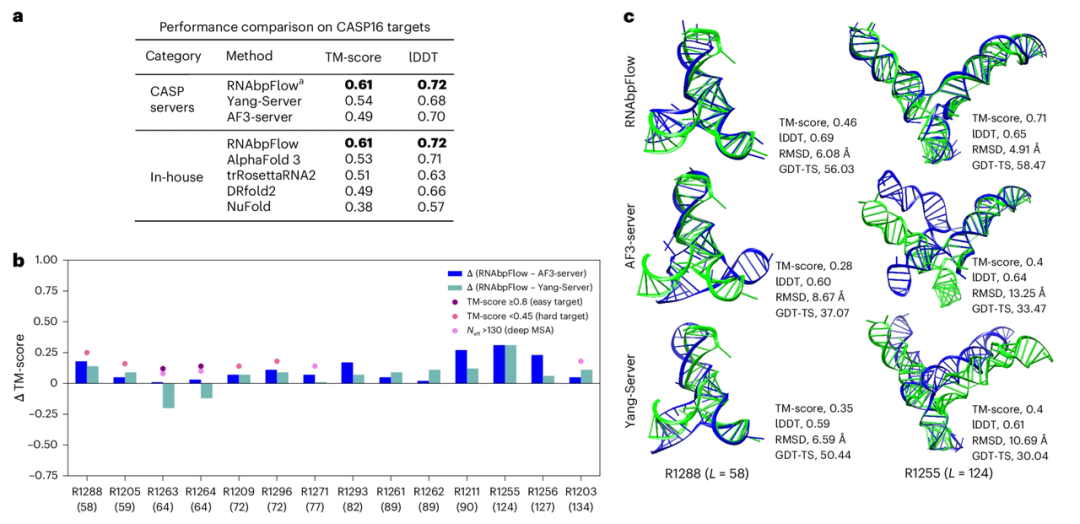
Furthermore, the experiments demonstrated that for unpredictable targets (TM-score < 0.45; MSA effective homology sequence depth ≤ 130; weak evolutionary signal), the structure prediction model based on base pairing constraints clearly outperformed RNAbpFlow when evolutionary information was scarce, showcasing the superiority of RNAbpFlow, which can predict the three-dimensional structure of RNA solely based on base pairing information. However, when facing easily predictable targets such as R1263 and R1264, sufficient deep multiple sequence alignment data enabled the two alignment models, AF3-server and Yang-Server, to be on par with or even better than RNAbpFlow, demonstrating the strong dependence of both models on sequence alignment information.
Final Thoughts
In summary, RNAbpFlow is not limited by MSA and structural homology. It can directly generate full-atom RNA three-dimensional structure models end-to-end using only sequence and base pairing. With the help of high-precision atomic-level large-scale conformation set generation technology, it may open up a very promising new direction for the study of RNA conformation dynamics.








